Quais os sintomas e causas do Angioedema Hereditário?
 |
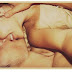
| Figura 1. (A) Radiografia abdominal do paciente e (B) massivo inchaço no cólon cirurgicamente exposto para fora da cavidade abdominal do paciente (foto sem censura, acesse aqui). Ref.1 |
Um homem de 37 anos de idade apresentou-se ao departamento de emergência com um histórico de 8 horas de dor severa e vômito. Uma radiografia abdominal revelou dilatação do intestino grosso com proeminentes alças no intestino delgado (Fig.1A). Exame com enema de bário revelou severa obstrução da flexura esplênica. Uma laparotomia de emergência foi realizada. O cólon transverso foi encontrado massivamente dilatado (Fig.1B) com severos edemas nas mucosas que estendiam proximalmente até o cólon ascendente. Uma resseção parcial do intestino grosso foi realizada. Após o procedimento, o paciente reportou um histórico de inexplicáveis episódios de dor abdominal e inchaço das suas mãos, pés e escroto, sem urticária ou coceira associada, desde os 18 anos de idade.
O paciente nunca recebeu tratamento com um inibidor da enzima conversora de angiotensina. Ele reportou que seu pai e parentes no lado paterno da família também manifestavam episódios similares. Testes laboratoriais subsequentes revelaram um baixo nível C4 e um nível de inibidor esterase C1 de 6 mg por decilitro (faixa de referência: 19 a 37 mg/dL). Um diagnóstico final de obstrução do intestino grosso causado por angioedema intestinal - no contexto de um diagnóstico atrasado de angioedema hereditário devido a deficiência do inibidor de esterase C1 - foi feito.
Tratamento profilático de longo prazo com danazol foi prescrito, junto com terapia de reposição do inibidor de C1-esterase para ataques recorrentes de angioedema, Além disso, o paciente foi submetido a um aconselhamento genético para a sua condição.
O relato de caso foi descrito e reportado no periódico The New England Journal of Medicine (Ref.1).
ANGIOEDEMA HEREDITÁRIO
O angioedema é um inchaço (edema) de início rápido, que se instala em um intervalo de tempo que varia de minutos a poucas horas e que acomete camadas profundas da pele, podendo estar associado à urticária. O inchaço é deformante, em geral assimétrico (compromete mais um lado do que o outro) e regride completamente (volta ao normal) após horas e dias. Acomete principalmente as pálpebras e lábios. Quando afeta a laringe, é conhecido popularmente como "edema de glote". Normalmente o angioedema não coça, mas pode causar dor e alteração de sensibilidade, parecida com uma "dormência".
Angioedema Hereditário (AEH) é uma doença genética, rara, potencialmente fatal e ainda pouco divulgada. A condição possui herança autossômica dominante, ou seja, os pais têm 50% de probabilidade de passar a alteração genética (mutação) para seus filhos. Não é uma alergia e sim um defeito do sistema imune (imunodeficiência), que resulta na produção exagerada de uma substância chamada bradicinina. Possui evidência estimada de aproximadamente 1 em cada 50000-100000 pessoas (Ref.3).
Caracteriza-se por crises de inchaço recorrente, grave, localizado, podendo ser desfigurante, acometendo não só a pele, como também órgãos internos, como a mucosa do intestino, levando a uma dor intensa e a aumento do volume abdominal. Durante uma mesma crise, o paciente pode apresentar edema em diferentes partes do corpo, por exemplo, inchar o braço e ter dor abdominal simultaneamente.
O Angioedema Hereditário (AEH) pode ser dividido em dois grupos:
Grupo 1. Com deficiência do inibidor de C1 (relacionado a mutações no gene SERPING1, que codifica o inibidor de C1):
- AEH Tipo I (80 a 85% dos casos) – produção insuficiente do inibidor de C1 com consequente deficiência quantitativa;
- AEH Tipo II (15 a 20% dos casos) – produção normal e defeito na função do inibidor de C1.
Grupo 2. Com inibidor de C1 normal (relacionado a mutações em outros genes):
- no gene F12, que codifica o fator XII (FXII) da coagulação;
- no gene da Angiopoietina 1 (ANGPT1);
- no gene do Plasminogênio (PLG);
- no gene do Cininogênio 1 (KNG1).
Existem situações onde, apesar de familiares serem acometidos, o gene responsável pela doença ainda não é conhecido. A AEH é causada primariamente por mutações no gene SERPING1 - e a maioria dos pacientes exibindo atividade do inibidor de C1 abaixo de 50% do normal -, com mutações em outros genes (Grupo 2) representando casos mais raros (pelo menos 10% do total).
-------------
> A doença [angioedema hereditário] tem esse nome por causa dos seus efeitos: alteração dos vasos sanguíneos (angio), causando inchaço (edema) e por ser transmitida geneticamente (hereditário).
> O inibidor de C1 (C1-INH), também denominado inibidor de C1-esterase, é uma importante proteína reguladora de diversos sistemas do organismo humano. Em condições normais, o inibidor de C1 regula estes sistemas, inibindo sua ativação excessiva. Quando ocorre a deficiência da quantidade e/ou da função do inibidor de C1, há uma ativação descontrolada desses sistemas e seus componentes (incluindo fator XIIa e calicreína no plasma), com consequente produção de substâncias pró-inflamatórias, principalmente bradicinina, resultando em vasodilatação, aumento da permeabilidade dos vasos e edema ("inchaço"). Mutações em outros componentes associados a esses sistemas regulados pelo inibidor de C1 podem também levar ao desenvolvimento de AEH, como substituições de lisina ou arginina no gene codificante do fator XIIa (Ref.5-6).
> É importante lembrar que um indivíduo pode desenvolver AEH sem qualquer um dos pais possuírem a doença. Mutações espontâneas nos genes associados à AEH podem emergir no desenvolvimento fetal ou durante as produção de gametas (gametogênese). É estimado que novas mutações representem 20-25% dos casos (Ref.7). Por isso, sintomas sugestivos de AEH devem ser investigados mesmo sem histórico familiar.
------------
O AEH caracteriza-se por crises recorrentes de edema na pele e/ou órgãos internos. Pode ocorrer em qualquer parte do corpo, mas em especial nas mãos, pés, face, pálpebras, boca, língua e órgãos genitais. É um inchaço que não coça, não dói nem deixa a pele vermelha ou empolada. As crises podem aparecer rapidamente ou evoluir em horas e persistir por 2 a 6 dias. Sintomas podem se manifestar em qualquer idade, mas >50% dos pacientes com AEH de Grupo 1 experienciam um ataque antes dos 18 anos de idade, como uma média de manifestação ao redor de 10 anos (Ref.8).
 |
| Figura 2. Duas pacientes (A-B) com inchaço progressivo (C-D) resultante de um ataque de angioedema. |
 |
| Figura 3. Ataques de angioedema em duas pacientes (A-B) com AEH, afetando o rosto (B-C) e a mão (B). Referência, acesse aqui. |
 |
| Figura 4. Paciente com AEH exibindo intenso inchaço no rosto durante um ataque de angioedema. Referência, acesse aqui. |
 |
| Figura 5. Uma paciente de 14 anos de idade com AEH durante ataque, exibindo inchaço espontâneo do lábio superior se estendendo até a linha mediana do lábio e região do pré-maxilar. Referência, acesse aqui. |
Alguns pacientes podem apresentar uma vermelhidão discreta na pele, descrita como eritema marginatum, que tem contornos sinuosos e centro claro; é mais comum em crianças (Fig.6). Este aspecto aparece antes de ocorrer o inchaço, sendo chamado de pródromo.
 |
| Figura 6. Eritema marginatum, um sinal cutâneo pródromo indicativo de um ataque de AEH. Afeta até 58% dos pacientes pediátricos com a condição. Pode se manifestar minutos até dias antes do desenvolvimento de inchaços no corpo, portanto facilitando reconhecimento precoce e prevenção. Ref.9 |
As crises podem afetar também a mucosa de órgãos internos, mais comumente intestinos e laringe. O edema das alças intestinais tem como sintomas dor abdominal, náuseas, vômitos, diarreia e/ou prisão de ventre. Casos graves podem evoluir para obstrução intestinal, simulando um abdômen agudo cirúrgico. Sintomas intestinais podem ser comumente confundidos com intolerâncias alimentares (ex.: glúten e lactose) (1) e apendicite. Não é incomum que sejam realizadas cirurgias desnecessárias, desde apendicectomia (remoção do apêndice) (2) até histerectomia (remoção do útero) (Ref.9). Quadros recorrentes e inexplicáveis de dor abdominal devem levantar suspeita para um quadro de AEH (Ref.9). Aliás, dor abdominal é muito comum em pacientes com AEH, ocorrendo em até 93% dos afetados pela condição, e quadros de dor abdominal recorrente em até 80% (Ref.10).
Leitura recomendada:
- (1) Forte seleção natural nos humanos para o consumo de leite na fase adulta
- (2) O apêndice intestinal é realmente um vestígio evolucionário inútil?
Quando a laringe é acometida (edema de glote), há risco de morte por asfixia. Se não tratados adequadamente, os registros mostram a possibilidade de desenvolver asfixia em 25-40% dos casos. Taxas de mortes por edema laríngeo ao redor do mundo possuem uma média de 1 para cada 20 pacientes (Ref.11). No geral, 30-60% das mortes por AEH estão associadas com edema laríngeo (Ref.11). Sintomas comuns podem incluir dificuldade para engolir, inchaço de toda boca (incluindo língua, céu da boca e úvula) e do pescoço, rouquidão e falta de ar. E importante realçar que manipulação da boca e da garganta deve ser evitada porque pode piorar o ataque (Ref.9).
Quando afeta a bexiga, retenção urinária é um sintoma comum.
Sinais e sintomas que falam CONTRA o diagnóstico de Angioedema Hereditário:
- Presença de coceira ou urticária;
- Melhora com antialérgico ou corticoide;
- Melhora muito rápida do edema, em menos de 24 horas.
-----------
> Evidências mais recentes também apontam que o AEH está associado com um risco maior de tromboembolismo venoso. Além disso, pacientes com AEH parecem exibir uma carga trombo-inflamatória elevada, podendo indicar que esses indivíduos estão continuamente em um estado subclínico de ataque fora de ataques clínicos com edemas. Ref.12-14
------------
As crises de AEH podem ser espontâneas ou provocadas por diversos fatores, tais como: emocionais (medo, perda de um ente querido), traumas (tratamentos dentários, cirurgias, procedimentos médicos), medicamentos, alterações hormonais, ingestão de alimentos, entre outros. Entretanto, nem sempre o fator que desencadeia o edema pode ser identificado, principalmente nas crianças. É ainda incerto como certos alimentos podem engatilhar ataques de AEH, com alguns estudos sugerindo hipersensibilidade IgE-mediada enquanto outros sugerem associação com uma reação de intolerância à histamina (Ref.9). A severidade da AEH pode ser agravada por outras desordens do trato gastrointestinal, como doença celíaca (3), doença de Crohn e infecção com a bactéria carcinogênica Helicobater pylori (4).
Leitura recomendada:
- (3) Cortar o glúten da dieta traz benefícios?
- (4) Importante lista de carcinógenos agora soma 256 substâncias
No AEH com inibidor de C1 normal, o desencadeante mais importante é o estrógeno (Ref.2). Por isso, este tipo de angioedema já foi "apelidado" de AEH estrógeno-dependente, visto que ocorre mais no sexo feminino. Algumas mulheres apenas desenvolvem a doença ao usar anticoncepcionais contendo estrógenos. É importante ressaltar que os mesmos fatores desencadeantes do AEH com deficiência do inibidor do C1 podem também podem ocasionar sintomas neste grupo.
Durante as crises, medicamentos comumente utilizados - e aprovados no Brasil -, são: concentrado do inibidor de C1 esterase humano e icatibanto. O primeiro é injetável (via venosa) e repõe o inibidor de C1, enzima deficiente no AEH com déficit do inibidor de C1. O icatibanto é um medicamento administrado pela via subcutânea (na gordura debaixo da pele) - preferencialmente na região abdominal - e bloqueia o receptor da bradicinina, que é a principal substância responsável pelo surgimento do edema.
O tratamento preventivo ou profilaxia de longo prazo no AEH é realizado para evitar o aparecimento das crises de inchaço, quando a doença não está controlada adequadamente. Neste caso, utilizam-se medicamentos diariamente, em doses determinadas pelo médico que acompanha o paciente. No Brasil, o tratamento é realizado com hormônios masculinos atenuados ou antifibrinolíticos, sob a forma de comprimidos. Vale lembrar que andrógenos e antifibrinolíticos não devem ser usados para tratamento de crises. Em outros países, o medicamento mais utilizado é o concentrado de inibidor de C1 injetável, sendo administrado de duas a três vezes por semana, por via venosa ou subcutânea. Existem também promissores medicamentos mais recentes sendo testados para uso subcutâneo que bloqueiam a calicreína (ex.: setralstat) ou inibem o fator XIIa (ex.; garadacimab), outros dois importantes mediadores no desenvolvimento do angioedema (Ref.15-16).
Um estudo clínico randomizado e placebo-controlado de Fase II publicado em 2022 no periódico NEJM (Ref.17), analisando um total de 20 pacientes com AEH causada por deficiência de inibidor de C1, mostrou que o medicamento donidalorsen - um inibidor de pré-calicreína no plasma - reduziu em quase 10 vezes a taxa mensal de ataques de angioedema, e com um bom perfil de segurança.
O tratamento preventivo ou profilaxia de curto prazo no AEH consiste no uso de medicamentos antes da realização de alguns procedimentos que possam desencadear crises de inchaço. Os tratamentos dentários, principalmente extração dentária, bem como aqueles que envolvem a manipulação da boca e garganta, tais como endoscopia digestiva alta, broncoscopia e intubação, são os que provocam crises com mais frequência. O concentrado do inibidor de C1 esterase por via venosa é indicado na profilaxia em curto prazo, uma hora antes do procedimento. Quando o concentrado não está disponível, utiliza-se o plasma fresco congelado, ou ainda, os hormônios masculinos atenuados
REFERÊNCIAS CIENTÍFICAS
- https://www.nejm.org/doi/full/10.1056/NEJMicm2303943
- https://asbai.org.br/wp-content/uploads/2019/11/projeto_Doutor_eutenho-angioedemaHereditario-v3.pdf
- Horiuchi & Miyata (2023). Biochemistry, molecular genetics, and clinical aspects of hereditary angioedema with and without C1 inhibitor deficiency. Allergology International, Volume 72, Issue 3, Pages 375-384. https://doi.org/10.1016/j.alit.2023.04.004
- https://ashpublications.org/blood/article/139/18/2816/483751/A-mechanism-for-hereditary-angioedema-caused-by-a
- Gailani et al. (2023). Mechanisms involved in hereditary angioedema with normal C1-inhibitor activity. Frontiers in Physiology, Volume 14. https://doi.org/10.3389/fphys.2023.1146834
- Bork et al. (2023). Gene Mutations Linked to Hereditary Angioedema in Solitary Angioedema Patients With Normal C1 Inhibitor. The Journal of Allergy and Clinical Immunology: In Practice, Volume 11, Issue 8, Pages 2441-2449. https://doi.org/10.1016/j.jaip.2023.01.051
- Prenzel et al. (2023). Epidemiology and treatment of children with hereditary angioedema in Germany: A retrospective database study. Clinical and Translational Allergy, Volume 13, Issue 11 e12313. https://doi.org/10.1002/clt2.12313
- Banerji et al. (2023). Hereditary Angioedema: A Review of the Current and Evolving Treatment Landscape. The Journal of Allergy and Clinical Immunology: In Practice, Volume 11, Issue 8, Pages 2315-2325. https://doi.org/10.1016/j.jaip.2023.04.017
- Magerl et al. (2023). Could it be hereditary angioedema?—Perspectives from different medical specialties. Clinical and Translational, Volume 13, Issue 9, e12297. https://doi.org/10.1002/clt2.12297
- Staller et al. (2022). Consider Hereditary Angioedema in the Differential Diagnosis for Unexplained Recurring Abdominal Pain. Journal of Clinical Gastroenterology, 56(9): 740–747. https://doi.org/10.1097%2FMCG.0000000000001744
- Minafra et al. (2022). The Mortality from Hereditary Angioedema Worldwide: a Review of the Real-World Data Literature. Clinical Reviews in Allergy & Immunology 62, pages232–239. https://doi.org/10.1007/s12016-021-08897-8
- Grover et al. (2022). Hereditary angioedema is associated with an increased risk of venous thromboembolism. Journal of Thrombosis and Haemostasis, Volume 20, Issue 11, P2703-2706. https://doi.org/10.1111/jth.15870
- https://ashpublications.org/blood/article/141/19/2295/495575/Hereditary-angioedema-and-thrombosis
- Gramstad et al. (2023). Increased thromboinflammatory load in hereditary angioedema, Clinical and Experimental Immunology, uxad091. https://doi.org/10.1093/cei/uxad091
- Aygören-Pürsün et a. (2023). An investigational oral plasma kallikrein inhibitor for on-demand treatment of hereditary angioedema: a two-part, randomised, double-blind, placebo-controlled, crossover phase 2 trial. The Lancet, Volume 401, Issue 10375, P458-463. https://doi.org/10.1016/S0140-6736(22)02406-0
- Craig et al. (2023). Efficacy and safety of garadacimab, a factor XIIa inhibitor for hereditary angioedema prevention (VANGUARD): a global, multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. The Lancet, Volume 401, Issue 10382, P1079-1090. https://doi.org/10.1016/S0140-6736(23)00350-1
- Lauré et al. (2022). Inhibition of Prekallikrein for Hereditary Angioedema. New England Journal of Medicina, 386:1026-1033. https://doi.org/10.1056/NEJMoa2109329