O que é a Doença Hepática Policística?
 |
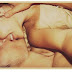
| Figura 1. Fígado removido da paciente ao lado do fígado doado para substituí-lo. Ref.1 |
Uma mulher de 51 anos idade com doença hepática e renal policística havia sido submetida a um transplante renal 21 anos antes da nova apresentação. Ela não apresentava evidências de malformações cerebrovasculares. Tanto seu pai quanto sua tia também tinham doença renal policística. Após o transplante renal, o fígado da paciente sofreu uma progressiva deterioração e aumento de volume (hepatomegalia) devido a alterações císticas.
Saciedade precoce, desnutrição e dor abdominal tornaram necessário um transplante de fígado. A massa corporal da receptora no momento do transplante era de 59 kg.
Um enorme fígado de 9,1 kg (flecha branca) foi removido e substituído por um enxerto íntegro que correspondia a um décimo do peso do fígado doente (flecha preta) (Fig.1). Um grande cisto na cúpula do fígado nativo precisou ser descompressado (ponta de seta branca) para permitir o acesso à veia cava supra-hepática da receptora (Fig.1).
Após o procedimento, a paciente teve uma excelente recuperação e apresentava função renal e hepática normais após 4 anos de acompanhamento.
O caso foi descrito e reportado em 2007 no periódico New England Journal of Medicine (Ref.1).
DOENÇA HEPÁTICA POLICÍSTICA
A doença hepática policística (DHP) é um distúrbio genético raro caracterizado por mutações em genes que codificam proteínas envolvidas no transporte de fluidos e no crescimento de células epiteliais no fígado. Essas mutações levam à substituição do tecido hepático normal por cistos hepáticos preenchidos por líquido. A condição é caracterizada por 10 ou mais cistos no parênquima do fígado (Ref.3).
Existem duas formas distintas de DHP: a DHP isolada e a DHP associada à doença renal policística (DRP).
Na DHP isolada, ocorrem mutações germinativas nos genes PRKCSH e SEC63. Esses genes codificam as proteínas sec-63 e hepatocistina, que atuam no transporte de fluidos e no crescimento das células epiteliais. Essas proteínas são expressas em hepatócitos e colangiócitos.
O segundo grupo de DHP compreende os casos associados à DRP. Existem duas formas de DRP: a DRP autossômica dominante e a DRP autossômica recessivA. Nessa forma de DHP, o comprometimento renal é predominante. Os pacientes tendem a desenvolver complicações renais, como insuficiência renal, sendo que a maioria necessita de diálise e transplante renal. Na DHP autossômica dominante, a causa primária são variantes patogênicas nos genes PKD1 (maioria dos casos) e PKD2 (Ref.5). Esses genes codificam a policistina 1 e a policistina 2, proteínas localizadas nos cílios de células epiteliais renais e que parecem mediar a contração do lúmen celular (Ref.6). A maioria dos pacientes com DHP autossômica dominante falece logo após o nascimento devido a complicações pulmonares. Aqueles que sobrevivem tendem a desenvolver fibrose hepática em vez de cistos.
----------
> Mais de 90% dos pacientes com mais de 35 anos de idade afetados pela DHP autossômica dominante exibem cistos hepáticos, e até 70% dos casos de doença hepática policística são causados pela condição. A DHP autossômica dominante é responsável por 5% a 10% das falhas renais nos EUA e na Europa. É a causa genética mais comum para doença crônica renal ao redor do mundo. Ref.5-8
> Mutações em múltiplos genes podem causar doença renal policística, mas os mais comumente envolvidos são o PKD1, PKD2 e PKHD1 - esse último responsável pela codificação da proteína fibrocistina. Ref.9
----------
A maioria dos pacientes com DHP é assintomática e recebe o diagnóstico incidentalmente por meio de exames de imagem.
A doença dificilmente interfere na fisiologia do fígado, o que confere um prognóstico razoável ao paciente, de acordo com o tamanho dos cistos e a sua interferência nas estruturas adjacentes. Apenas 10-20% dos pacientes apresentam manifestações clínicas resultantes da hepatomegalia - essa última causada pelos múltiplos cistos (Ref.10). Um fígado normal adulto possui em torno de 1,5 kg, mas o fígado de alguns pacientes com DHP pode superar 10 kg de massa acumulada. Na média, o fígado policístico aumenta até ~6,7 kg, com uma taxa de crescimento anual de até 3,7% (Ref.11-12).
 |
| Figura 2. Paciente de 57 anos de idade do sexo feminino com DHP autossômica dominante e doença hepática policística. O fígado policístico da paciente acumulou uma massa de 14 kg. Devido a vários sintomas debilitantes e perda de peso severa causados pelo grande efeito de massa, a paciente precisou ser submetida a um transplante de fígado. Em (a), tomografia computadorizada em corte axial mostrando a anatomia abdominal, com o rim (K) e o pâncreas (P) em relação ao fígado. Ref.12 |
Nos pacientes sintomáticos, a hepatomegalia pode causar dor abdominal, distensão e compressão de órgãos adjacentes, fadiga e dispneia, afetando potencialmente a qualidade de vida. Para pacientes com DHP sintomática, o objetivo terapêutico principal é reduzir o volume hepático. Casos muito severos podem requerer cirurgia, incluindo fenestração de cistos, ressecção hepática e transplante de fígado.
O diagnóstico da DHP pode ser realizado por meio de ultrassonografia, tomografia computadorizada (TC) ou ressonância magnética (MRI).
Leitura recomendada:
REFERÊNCIAS
- William J. Wall (2007). Liver Transplantation for Polycystic Liver Disease. NEJM, 356;15. https://www.nejm.org/doi/full/10.1056/NEJMicm055470
- https://www.ncbi.nlm.nih.gov/books/NBK549882/
- https://britishlivertrust.org.uk/information-and-support/liver-conditions/polycystic-liver-disease/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC8987261/
- Chebib et al. (2025). Autosomal Dominant Polycystic Kidney Disease A Review. JAMA Network, 333;(19):1708-1719. https://doi.org/10.1001/jama.2025.0310
- Ma et al. (2023). Polycystic kidney disease: novel insights into polycystin function. Trends in Molecular Medicine, Volume 29, Issue 4, P268-281. https://doi.org/10.1016/j.molmed.2023.01.005
- Pei et al. (2026). Autosomal dominant polycystic kidney disease. The Lancet, Volume 407, Issue 10535, P129-1302. https://doi.org/10.1016/S0140-6736(26)00046-2
- https://my.clevelandclinic.org/health/diseases/polycystic-liver-disease
- Boletta & Caplan (2024). Physiologic mechanisms underlying polycystic kidney disease. Physiological Reviews, Volume 105, Issue 3. https://doi.org/10.1152/physrev.00018.20
- Neto et al. (2023). Combined liver-kidney transplant in polycystic diseases: a case report. Einstein (São Paulo), 21. https://doi.org/10.31744/einstein_journal/2023RC0282
- https://pmc.ncbi.nlm.nih.gov/articles/PMC5816892/
- Desai et al. (2018). Liver Transplant for Unusually Large Polycystic Liver Disease: Challenges and Pitfalls. Case Reports in Transplantation. https://doi.org/10.1155/2018/4863187